The Consortium of Molecular Design at BYU provides cutting edge interdisciplinary research opportunities for students to push the envelope for protein engineering and drug discovery.
We use close collaboration between laboratories at BYU in Physics, Chemistry, Computer Science, LifeSciences, and Engineering to tackle these challenging topics from all angles.
We actively seek industrial collaboration and support for our efforts and are excited to explore mutually beneficial application of all state-of-the-art technologies to revolutionize molecular design.
News and Events
Selected Publications

Background
The quality of carbohydrate intake, as measured by the glycemic index (GI), has not been evaluated nationally over the past two decades in the United States (US).
Objective
We aimed to develop a comprehensive and nationally representative dietary GI and glycemic load (GL) database from 1999-2018 National Health and Nutrition Examination Survey (NHANES) and to examine GI and GL time trends and sub-population differences.
Design
We employed an artificial intelligence (AI)-enabled model to match GI values from two GI databases to food codes from US Department of Agriculture, which were manually validated. We examined nationally representative distributions of dietary GI and GL from 1999-2018 using the multistage, clustered sampling design of NHANES.
Results
Assigned GI values covered 99.9% of total carbohydrate intake. The initial AI accuracy was 75.0%, with 31.3% retained after manual curation guided by substantive domain expertise. A total of 7,976 unique food codes were matched to GI values, of which soft drinks and white bread were top contributors to dietary GI and GL. Of the 49,205 NHANES adult participants, the mean dietary GI was 55.7 [95% CI: 55.5, 55.8] and energy-adjusted dietary GL was 133.0 [132.3, 133.8]. From 1999 to 2018, dietary GI and GL decreased by 4.6% and 13.8%, respectively. Dietary GL was higher among females 134.6 [133.8, 135.5] than males 131.3 [130.3, 132.3], those with ≤ high school degree 137.7 [136.8, 138.7] compared to those with ≥ college degree 126.5 [125.3, 127.7], and those living under the poverty level 140.9 [139.6, 142.1] compared to above. Differences in race were observed (Black adults 139.4 [138.2, 140.7]; White adults 131.6 [130.5, 132.6]).
Conclusion
We developed a national GI and GL database to facilitate large-scale and high-quality surveillance or cohort studies of diet and health outcomes in the US.
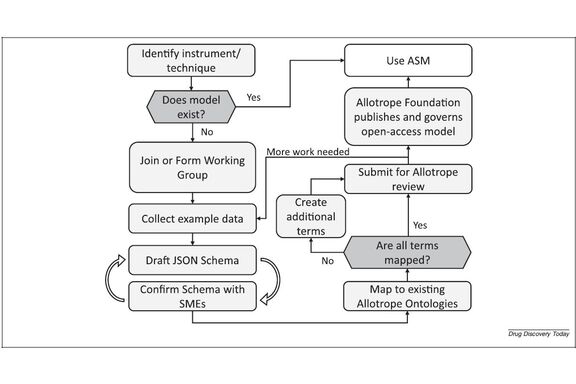
The Allotrope Foundation (AF) started as a group of pharmaceutical companies, instrument, and software vendors that set out to simplify the exchange of data in the laboratory. After a decade of work, they released products that have found adoption in various companies. Most recently, the Allotrope Simple Model (ASM) was developed to speed up and widen the adoption. As a result, the Foundation has recently added chemical companies and, importantly, is reworking its business model to lower the entry barrier for smaller companies. Here, we present the proceedings from the Allotrope Connect Fall 2023 conference and summarize the technical and organizational developments at the Foundation since 2020.

Accurate interatomic energies and forces enable high-quality molecular dynamics simulations, torsion scans, potential energy surface mappings, and geometry optimizations. Machine learning algorithms have enabled rapid estimates of the energies and forces with high accuracy. Further development of machine learning algorithms holds promise for producing universal potentials that support many different atomic species. We present the Transformer Interatomic Potential (TrIP): a chemically sound potential based on the SE(3)-Transformer. TrIP’s species-agnostic architecture, which uses continuous atomic representation and homogeneous graph convolutions, encourages parameter sharing between atomic species for more general representations of chemical environments, maintains a reasonable number of parameters, serves as a form of regularization, and is a step toward accurate universal interatomic potentials. TrIP achieves state-of-the-art accuracies on the COMP6 benchmark with an energy prediction of just 1.02 kcal/mol MAE. We introduce physical bias in the form of Ziegler–Biersack–Littmark-screened nuclear repulsion and constrained atomization energies. An energy scan of a water molecule demonstrates that these changes improve long- and near-range interactions compared to other neural network potentials. TrIP also demonstrates stability in molecular dynamics simulations, demonstrating reasonable exploration of Ramachandran space.